citation and acknowledgements
We're now published in Bioinformatics:
ARResT/AssignSubsets: a novel application for robust subclassification of chronic lymphocytic leukemia based on B cell receptor IG stereotypy
Vojtech Bystry; Andreas Agathangelidis; Vasilis Bikos; Lesley Ann Sutton; Panagiotis Baliakas; Anastasia Hadzidimitriou; Kostas Stamatopoulos; Nikos Darzentas
Bioinformatics 2015
doi: 10.1093/bioinformatics/btv456
| acknowledgements
The tool is currently supported by Ministry of Health of the Czech Republic grant nr. 16-34272A.
news and updates
07.01.22 | fix for IMGT switching to https
10.11.19 | fix for sequence labels with tabs
- rerun sequences if you suspect this, saw "no (healthy) data to work with, please check the ARResT/SeqCure report", and sequence looked OK
28.08.19 | fix for IMGT/V-QUEST 3.5.8
26.07.19 | quick response from IMGT, we should be back to normal
26.07.19 | temporary fix for IMGT/V-QUEST 3.5.3 and its modified API; we're missing IMGT's run parameters, shouldn't affect results, but report any issues
29.05.19 | update for IMGT/V-QUEST 3.5.0 and its new API; shouldn't affect results, but please report any issues
11.10.18 | fix in ARResT/SeqCure
29.06.18 | improvements in ARResT/SeqCure, esp. for problematic sequences and feedback to user (thanks to Elisa Pagnin for the motivation)
08.02.18 | 'plain-text-formatted results table' now contains the full nucleotide sequence plus IMGT/V-QUEST metadata (e.g. versions and date);
plus code optimisations and cosmetic changes; please report any issues you might encounter
04.02.18 | model with latest IMGT/V-QUEST annotations, on which our update tests and stats are identical to previous ones,
but right-on-the-border 'borderline' user cases might now be assigned differently - if in doubt, please rerun such cases;
also, underlying code has been rewritten and software (e.g. R) updated, so please report any issues you might encounter
finally, we now link subsets assignments to ARResT/Subsets, the Encyclopedia of CLL Subsets
17.07.17 | functionality and its comments were not properly used in ARResT/SeqCure due to header changes in IMGT/V-QUEST 3.4.6 (30 March 2017)
- subset assignment NOT affected
26.02.17 | a bit more important textual information on assignment confidence levels (see DISCLAIMER, the 'score' section below, and the results page)
05.10.16 | a couple of output fixes (ordering of unassigned, and printing of unassigned when all scores were -Inf) - actual results NOT affected
04.10.16 | more user-friendly results ordering: first by assignment or not (naturally, and as was), then confidence (this is new),
then assignment score margin (not visible by itself thus confusing because it is subset-specific and was ordering 'confidence' unnaturally)
05.03.16 | now reporting comparative sequence issues, e.g. identical labels across sequences - ARResT/SeqCure could always do that, now here as well
10.10.15 | timeout issue with long runs should be resolved - apologies for any downtime or erroneous ("no (healthy) data...") runs, please repeat if unsure
09.08.15 | ARResT/AssignSubsets now published in Bioinformatics - with many thanks to the anonymous reviewers and the editorial staff
21.06.15 | earlier check for whether IMGT is accessible, and proper message when it's not, like (apparently) this weekend
21.06.15 | addition of bits of text to the output to make it easier to spot the links to the heat maps
04.06.15 | improvements in instructions, HTML formatting, and overall usability
16.03.15 | online
background and instructions
| intro
ARResT/AssignSubsets was built to robustly assign user-submitted sequences as new members to existing subsets of stereotyped antigen receptor sequences - that implies that ARResT/AssignSubsets does not discover new subsets. This is currently applicable to the 19 major subsets of stereotyped B-cell receptors in CLL. Critically, major subsets account for >10% of the cohort, >40% of stereotyped cases, and >25% of patients requiring treatment, and seem to share clinicobiological features and, even, outcome.
The tool is currently supported by Ministry of Health of the Czech Republic grant nr. 16-34272A.
| sequence features used in curation, learning and assignment
Immunogenetic annotations of sequences are obtained from IMGT/V-QUEST, the widely-accepted reference for antigen receptor sequence analysis. Initially, and importantly, these are used by ARResT/SeqCure to report on issues with the sequences which could potentially compromise the analysis.
The annotations are then used to extract sequence features, which are essential for both the learning and assignment phases of ARResT/AssignSubsets, and which are divided into two groups: core and secondary.
Core features are strictly controlled: if the value of a core feature for a given new sequence is not shared amongst the members of a given subset, then the sequence cannot be assigned to that subset. These are:
1. VH CDR3 length, a critical determinant of the structure of the antigen recognition loop;
2. IGHV gene phylogenetic clan, exploiting the sequence similarity of IGHV genes originating from the same clan; and
3. mutational status of the rearrangement, with 'mutated' for less than 98% nucleotide identity to germline and 'unmutated' otherwise, an important prognostic indicator in patients with CLL.
Secondary features are not critical and therefore unobserved values are accepted, but they do affect the subset assignment based on their respective values and frequencies in each major subset. These are:
1. relative frequency of IGHV and IGHJ genes, especially relevant in subsets with rearrangements that express more than one IGHV gene (prime example is subset #1) and in which not all IGHV genes are equally represented;
2. frequency of each amino acid at any given position of the VH CDR3 - e.g. the presence at a VH CDR3 position of an amino acid that is highly frequent among the sequences of a subset will increase the score, and thus the possibility that the rearrangement will be assigned to this subset.
| learning and assignment
ARResT/AssignSubsets has two major phases to serve its purpose:
1. learning is achieved through sets of rules captured in a probabilistic model, specifically a simple Bayesian network implemented in R [r-project.org/];
2. assignment is based on the abundance and statistical significance of evaluated sequence features, the same ones used to build the Bayesian network in the learning phase.
| score
The score is the difference between the best-scoring major subset and a negative set ('pool' cohort) of sequences belonging to minor subsets as well as non-subset, heterogeneous cases. Positive numbers mean the input sequence is closer to the subset, while negative numbers mean the input sequence is closer to the 'pool' cohort. '-Inf' (minus infinity) meaning that the core features of the submitted sequence did not match the core features of the subset. Assignment is based on a per-subset threshold, in turn based on the range of scores achieved by existing members of that subset. Finally, the difference between the relative score and the respective subset threshold is 'translated' to assignment confidence, ranging from 'borderline' to 'extreme', to help the user better evaluate the results. Our current results indicate ~4% of 'borderline' and 'low' assignments (essentially outlier members), whereas 'high' and 'extreme' take up ~90% with 'average' another ~5%. Therefore, and although low levels of confidence might still represent true assignments, users are urged to use caution and their best judgement or ask for advice in those cases, especially in a clinical context.
| input format
Provide up to 50 nucleotide sequences of immunoglobulin (IG) gene rearrangements in FASTA format.
We also offer an example set, just click 'FASTA' under 'or click to load example'.
| output
Note that there is also quick help in the results page, under 'click to open/close quick help'.
general output
After you launch ARResT/AssignSubsets, you'll be kept updated and presented with information and help, results and tables - please click on the screenshot below to get a larger version, and read below it for an explanation of all the different highlighted sections:
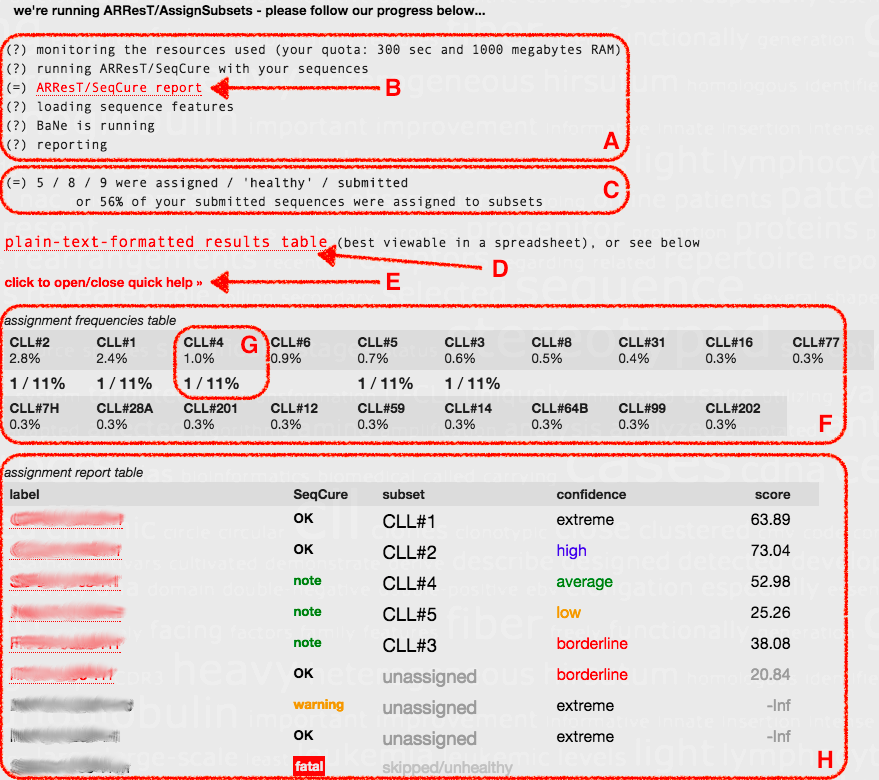
There are A-H sections on the screenshot - these are:
A. Real-time progress update, which includes
B. a detailed ARResT/SeqCure report, to assess the 'health' of your sequences - please don't ignore this information, you might compromise your analysis.
C. How many of your sequences were assigned to subsets, how many were 'healthy' according to ARResT/SeqCure, and how many you submitted.
D. Plain-text-formatted results table, amenable to (down)loading to Excel, with IMGT/V-QUEST annotation.
E. Interactive link to show/hide some extra summary information about the two following tables (F and H).
F. Assignment frequencies table for all subsets, e.g.
G. from top to bottom, the subset name, its relative frequency in the original published cohort, and how many of the submitted sequences were assigned to it
H. Assignment report table, which contains an overview of the results, including links to heat maps with core and secondary features and their contribution to and significance in the assignment process. In more detail, and regarding columns from left to right:
'label' - label of your sequence, also and when applicable linked to a heat map visualisation of the assignment result (more on heat maps below)
'SeqCure' - colour-coded 'worst' message level from ARResT/SeqCure, with 'fatal' leading to the sequence not being used for assignment
'subset' - subset your sequence was assigned to, or 'unassigned' otherwise, or 'skipped/unhealthy' when the sequence was not used
'confidence' - (non-)assignment confidence, as colour-coded 'borderline/low/average/high/extreme' (see 'score' above for more)
'score' - difference between scores to best-scoring subset and 'pool' cohort, >0 closer to subset, <0 closer to 'pool', -Inf: core features not found
heat maps
As promised, some more information on heat maps. Firstly a generic example with generic insight:
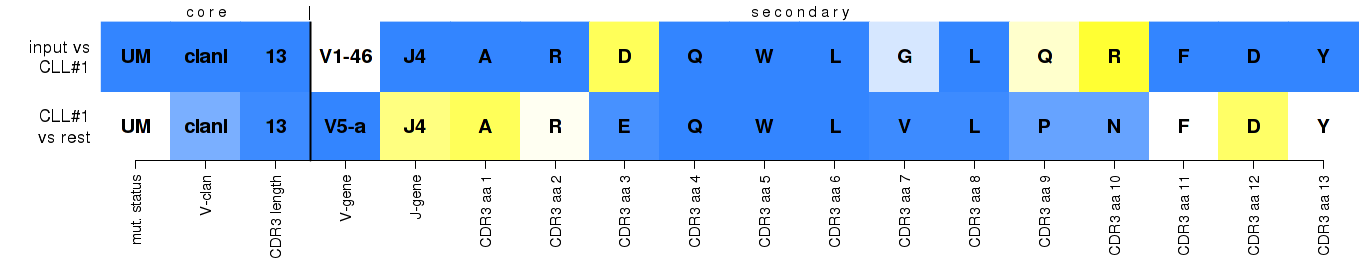 yellow-to-white-to-blue color scale...
yellow-to-white-to-blue color scale...
top row: increasing feature support within the assigned subset
bottom row: increasing feature uniqueness across all subsets, essentially highlighting subset-distinctive features
Two-row heat maps display the core and secondary sequence features that were evaluated, and their contributions and significance for assigning select IG gene rearrangements to major CLL subsets. The top row refers to the submitted rearrangement, and the bottom row to the theoretical rearrangement that would have achieved the highest score for every feature in the respective subset - its 'best member'. Regarding the yellow-to-white-to-blue color scale: for the submitted rearrangement on the top row, it reflects the support of the observed values against the support for the values of the 'best member' within the assigned subset; for the 'best member' on the bottom row, it reflects the increasing level of uniqueness of the value within the assigned subset against the 'pool' cohort, thus essentially highlighting subset-distinctive features. Below you'll find some more specific examples, with more detailed explanations about what you see and why, and how to possibly interpret similar results.
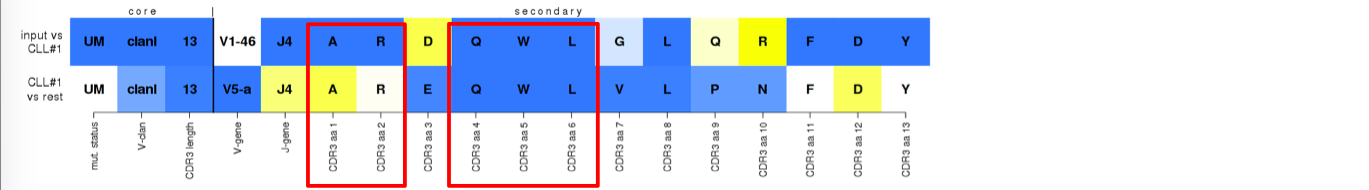
^True assignment to subset #1. The QWL pattern (VH CDR3 aa positions 4-6), previously shown (and confirmed herein) to be highly popular in (deep blue in top row) and specific to (deep blue in bottom row) subset #1, is preserved in the submitted sequence. In contrast, Alanine (A) and Arginine (R) residues at VH CDR3 aa positions 1 and 2 (IMGT 105-106) are also highly popular (deep blue in top row) but not equally distinctive (white in bottom row).
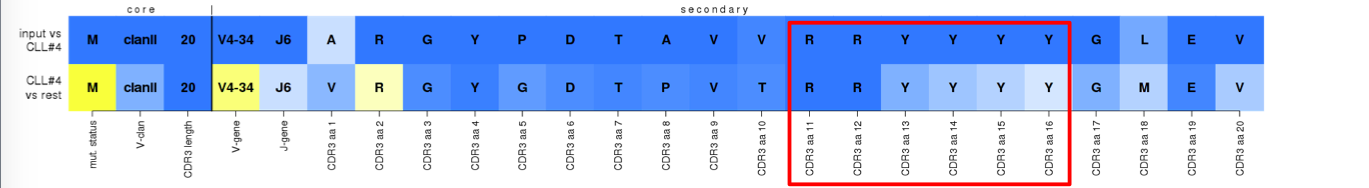
^True assignment to subset #4. Although the widely-used IGHJ6 tetrapeptide YYYY (VH CDR3 aa positions 13-16) is not subset-#4-specific as revealed by the light blue colors on the bottom row, joint coding (especially of the first two Y) with the 'junctional' dipeptide RR (VH CDR3 aa positions 11-12) makes it a deeply preserved and distinctive feature.
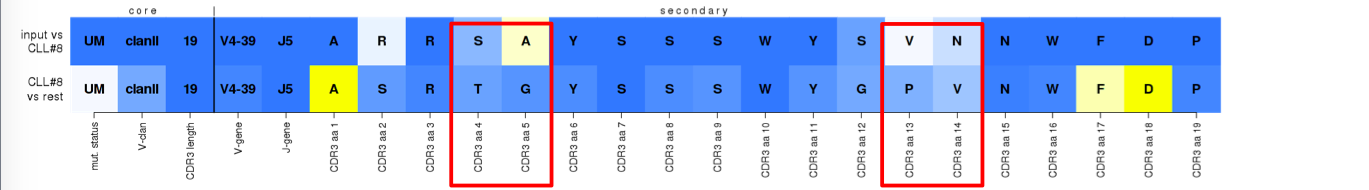
^True assignment to subset #8. An example of a 'mainly-germline' type of stereotypy, implied by the lighter blue color of the majority of VH CDR3 positions on the bottom row. The assigned sequence follows most of the 'best-member' features with few exceptions in the apparently 'junctional' VH CDR3 positions.
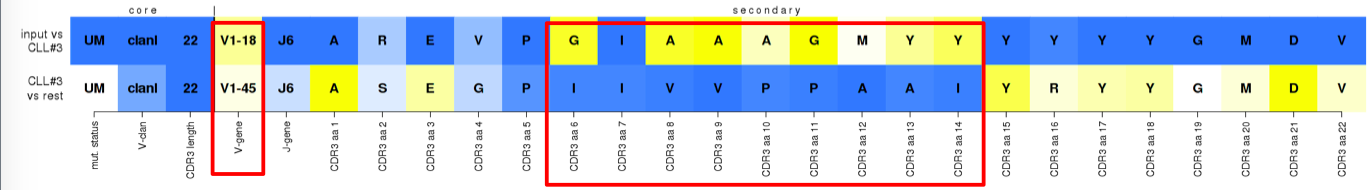
^False assignment to subset #3. Nearly all distinctive features of the subset, e.g. the IGHV gene and VH CDR3 aa positions 6-14 (IMGT 110-112.3), are replaced by infrequently observed, i.e. less supported, values in the submitted sequence. Nevertheless, the preservation of the core features as well as other secondary ones, falsely placed this sequence above the assignment threshold.
ARResT/SeqCure
Finally, some orientation around the ARResT/SeqCure report. The few sequences partially shown are the same as for the screenshots above.

A. Basic immunogenetic annotation from IMGT/V-QUEST for the sequence (label on the left hand side), including length in nucleotides, IMGT-Junction amino acid sequence, IGHV gene assignment(s), % identity to germline, etc
B. Issues discovered by ARResT/SeqCure through IMGT/V-QUEST annotation and application of expert rules, including a colour-coded message level (the same you see in ARResT/AssignSubsets' 'assignment report table' (H above)). Note that this sequence had a potential indel which guided ARResT/SeqCure to rerun IMGT/V-QUEST with its indel mode activated on to confirm it, which it did.
C. This sequence had an 'n' character which led to its exclusion from the rest of the analysis - here you see the specific message.
D. Advice regarding the issue from C above.
E. Full advice regarding the issue from C above, expanding on D.
hosted at the Bioinformatics Analysis Team / BAT